-
Apps and Services
-
LiteMol
A powerful 3D molecular viewer -
PatternQuery
Find atom patterns in molecular databases -
CoordinateServer
Streaming of 3D molecular data -
DensityServer
Streaming of 3D experimental data -
AtomicChargeCalculator II
Compute, compare, and visualize atomic charges -
MotiveValidator
Validate residues, ligands, and molecular fragments -
ValidatorDB
PDB database validated using MotiveValidator -
ValTrendsDB
Discover trends in structure quality -
MOLE 2.5
Compute and analyze channels -
CrocoBLAST
Local sequence alignment
-
- Wiki
- About
AtomicChargeCalculator
Conformationally Dependent Atomic Charges of Quantum Mechanics Quality
×
Atomic Charge Calculator II is now available!
AtomicChargeCalculator (ACC) is a complex yet intuitive utility for the calculation, visualization and analysis of atomic charges for small druglike molecules, as well as for large biomolecular complexes. Up to thousands molecules can be processed in one run. Empirical atomic charges that respond to changes in molecular conformation are calculated via an efficient implementation of the well established Electronegativity Equalization Method (EEM). ACC allows to use any of the EEM parameters sets published in literature. You may download your results, or visualize and analyze them directly in the web browser.
×
What can I do with atomic charges? Find out here.
This is the ACC Submission Page, where you can setup your calculations or check out useful examples. The results of your calculation will be available at a unique URL, which you may access at a later time. The results are organized on two levels. The ACC Synopsis page provides an overview of the results for all molecules, whereas the ACC Specifics page allows to investigate the results for each molecule separately.
The various sections of the web page offer interactive guides indicated by which give a quick walk through all the main elements of the page. Further help is provided by the info icons . Many tool tips are available by hovering over any graphical or textual element in the interface. Refer to the Wiki for any clarifications.
×
First Time User?
Go through the First Time User Presentation, or check out the
at the top right corner of the page.
If you found AtomicChargeCalculator helpful, please cite:
Ionescu,C.-M., Sehnal,D., Falginella,F.L., Pant,P., Pravda,L., Bouchal,T., Vařeková,R.S., Geidl,S. and and Koča,J. (2015)
AtomicChargeCalculator: Interactive Web-based calculation of atomic charges in large biomolecular complexes and drug like molecules. J. Cheminform., 7, 50.

AtomicChargeCalculator is a part of services provided by ELIXIR –
European research infrastructure for biological information.
For other services provided by ELIXIR's Czech Republic Node visit www.elixir-czech.cz/services.
License
Licence conditions in accordance with § 11 of Act No. 130/2002 Coll. The owner of the software is Masaryk University, a public university, ID: 00216224. Masaryk University allows other companies and individuals to use this software free of charge and without territorial restrictions in usual way, that does not depreciate its value. This permission is granted for the duration of property rights. This software is not subject to special information treatment according to Act No. 412/2005 Coll., as amended. In case that a person who will use the software under this licence offer violates the licence terms, the permission to use the software terminates.
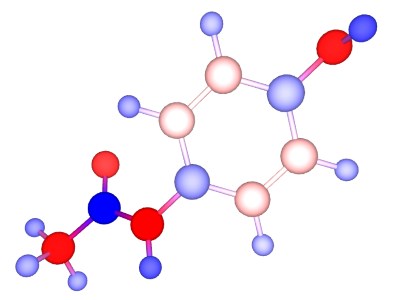
Paracetamol Drug-like molecule Browse Data
N-acetyl-p-aminophenol, commonly known as paracetamol, is a widely used analgesic and antipyretic. Its mechanism of action is believed to be the inhibition of the protein cyclooxygenase 2, regulating the production of pro-inflamatory compounds. The metabolic break down of paracetamol has been the subject of intense study, since it holds the key to both its therapeutic action and toxicity... [show more]
We have computed atomic charges in paracetamol (ideal model coordinates from wwPDB CCD entry TYL). The default ACC settings were used. Specifically, the total molecular charge was 0. The EEM parameter set Ouy2009_elem covered the NPA charge definition at the B3LYP/6-31G* level of theory, and was developed for small molecules. NPA charges generally support qualitative chemical concepts like induction and conjugation. The full EEM matrix was solved in double precision. The computation took less than 1s.
A quick analysis (e.g., sorting according to atomic charge value in the Raw Data tab) reveals that the phenolic H (position HO4) is the most acidic proton (highest positive charge) in the molecule. This suggests a faster and easier dissociation of this O-H bond. Indeed, up to 90% of metabolic degradation happens at position HO4 (glucuronidation, sulphonation). Additionally, up to 15% of metabolic degradation involves oxidation at the phenolic (HO4) and amidic positions (HN), the two most positive H in the paracetamol molecule. While paracetamol is a very small molecule with few polar sites, the same principle can be applied in reasoning out highly reactive sites in more complex molecules.
In general, it is a good idea to try several charge definitions. One or more of these might prove useful in further modeling studies. If you plan to run molecular dynamics, it is good to use charges which are compatible with the particular force field you plan to use. For instance, if the force field is known to use atomic charges based on electrostatic potential mapping, pick at least one EEM parameter set developed with this charge definition.
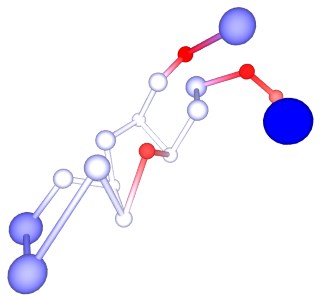
Protegrin-1 Antimicrobial peptide Browse Data
Protegrins are a family of antimicrobial peptides active against a wide range of pathogens. Protegrin-1 (PG1) has been intensely studied for its potential in treating infections caused by antibiotic resistant bacteria. PG1 shows activity against several pathogens, but also toxicity against the host. Useful mutations are those which maintain the antimicrobial activity, and at the same time reduce toxicity... [show more]
We calculated atomic charges in PG1 using ACC (coordinates from NMR model 1 of the PDB entry 1PG1). The total molecular charge was +7, owing to the many ARG residues. The EEM parameter set used was EX-NPA_6-31Gd_gas, which covered the NPA charge definition at the HF/6-31G* level of theory, and was developed for proteins. However, it was necessary to add to this set EEM parameters for deuterium (D), because this element was present in the input file. The EEM parameters for D were identical to the parameters for H. The full EEM matrix was solved in double precision. The computation took less than 1s.
PG1 contains 18 residues, and rather than analyzing atomic charges, we analyzed the residue charges, which are also reported by ACC (in all tabs, select the grouping Residues). PG1 is special because of its high positive charge. It contains 6 ARG residues. However, not all have the same charge. In particular, ARG at positions 4 and 9 have the least positive charge (around +0.5e), whereas the rest have much higher positive charge (over +0.8e). Such results suggest that mutations of ARG into a neutral residue at positions 4 or 9 would have a lower effect than mutations at positions 1, 10, 11 or 18.
A quick literature search reveals that many mutants of PG1 have been studied. Indeed, Ostberg and Kastnessis found that the mutation of ARG 4 into a neutral residue alters the antimicrobial activity against C. albicans significantly less than the mutation of ARG 10. Such biologically relevant insight can be gained by analyzing the residue charges on a single structure of PG1.
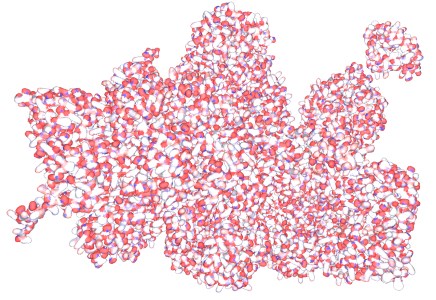
Proteasome Biomacromolecular complex Browse Data
The 26S proteasome is a large biomacromolecular complex which facilitates the targeted degradation of intracellular proteins, and thus plays an essential role in keeping protein homeostasis. It consists of a catalytic particle, made up of alpha rings and beta rings, controlled by regulatory particles, which are made up of a number of proteins. The proteasome is an intricate molecular machine which requires complex regulation to unfold and deubiquitilate the substrate, and push it through the catalytic machinery... [show more]
Necessarily, the proteasome undergoes large conformational changes during its operation. However, due to its size, such changes are very difficult to study. Recent work by Unverdorben et. al in the field of cryo-electron microscopy has lead to the discovery of intermediate conformers during the initial binding of ubiquitylated substrates. While the conformational changes in the regulatory particle are easily distinguishable, the changes in the catalytic particle are very subtle, due to the fact that all studied conformers refer to the initial phase of substrate binding.
Using ACC, we calculated atomic charges in the 3 conformers of the 26S proteasome that are available in the Protein Data Bank (PDB IDs 4CR2, 4CR3, 4CR4). The default ACC settings were used. Specifically, the total molecular charge was considered neutral for all conformers. The EEM parameter set used was EX-NPA_6-31Gd_gas, which covered the NPA charge definition at the HF/6-31G* level of theory, and was developed for proteins. The computation method was EEM Cutoff Cover, with a cutoff radius of 12Å. The computation took 130s.
The calculation produced 3 sets of atomic charges, one for each conformer. Since the proteasome is very large, we analyzed the residue charges, which are also reported by ACC, and subsequently the charges for the various subunits that make up the proteasome. Note that information about the subunits is given in the COMPND records of the PDB entries used here. Residues with the chain identifier 1-7 correspond to subunits beta 1-7. Residues with the chain identifier A-G correspond to subunits alpha 1-7. Finally, residues with the chain identifier H-Z correspond to regulatory subunits. Therefore, the total charge for a specific subunit or particle can be obtained as the sum of the charges for the residues with the corresponding chain identifiers. We thus followed the change in total charge for the regulatory particle, the catalytic particle, then separately for the alpha and beta rings.
The first observation was that, during the conformational changes from state 1, to state 2, and then to state 3, a significant amount of electron density is transferred between the catalytic particle and the regulatory particle. This suggests that, even though there is no significant movement observed for the catalytic particle, allosteric information is exchanged with the catalytic particle, and this information can be tracked at the electrostatic level. The next observation is that significant information is disseminated not only horizontally (within the alpha ring, or within the beta ring), but also vertically. In the overall transition it appears that the alpha ring loses electron density to the regulatory particle. By checking the intermediate state 2 it is possible to see that there is also transfer between the alpha ring and beta ring. This vertical shuttling of electron density within the catalytic particle suggests that the activity of alpha and beta subunits may cross-correlate. Such phenomena have indeed been reported.
For example, O'Hara et. al found that subunits alpha5 and beta1 may translocate together, while Cron et. al found that knockdown of alpha1 leads to loss of chymotrypsin activity associated with beta5. Further analysis can even yield the residues involved in the allosteric regulation, as those residues which exhibit a high variation in total charge (e.g., approximately 10 sites on the Rpn-13 regulatory subunit). This case study shows how a brief calculation using only a crude structural approximation can give insight regarding allosteric regulation in large biomolecular complexes.
Select a single file or a ZIP file containing molecule(s) in supported format (50MB limit).
Please ensure the molecules are complete and in a relevant conformation before uploading them.
See this page for complete info about how to prepare input files.
See this page for complete info about how to prepare input files.
Service version 1.0.23.12.27b (change log).
Command line version of the ChargeCalculator service
The command line version provides access to the underlying service. For usage instructions, please consult the
Wiki page.
Current Version (1.0.23.12.27b)
All Versions (change log):
Download